
Simulating Biomolecules with Python
| Category: | Science |
|---|---|
| Keywords: | Data Visualization, Biology, Computational Chemistry |
| Title: | Simulating Biomolecules with Python |
| Author: | Konrad Hinsen |
| Date: | 2005-04-20 |
| Website: | http://dirac.cnrs-orleans.fr/MMTK/ |
| Summary: | Python and C serve as the basis for a molecular modeling toolkit. |
| Logo: |
Background
The Molecular Modeling Toolkit (MMTK) is a open source Python library for molecular modeling and simulation with a focus on biomolecular systems, written in a mixture of Python and C. It provides standard techniques such as Molecular Dynamics or normal mode calculations in a ready-to-use form, but also provides a basis of low-level operations on top of which new techniques can easily be implemented.
I started developing MMTK in 1996. I had some experience with mainstream simulation packages for biomolecules that were written in Fortran and had their origins in the 1970s. Those packages were too cumbersome to use and in particular to modify and extend. Since my research work is focused on the development of new simulation techniques, modifiability was a particularly important criterion.
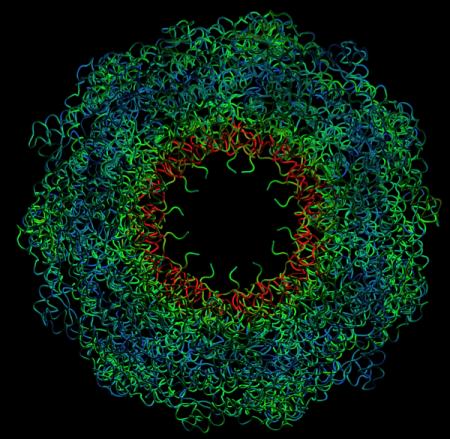
Dynamic deformation of the chaperon protein GroEL, obtained with the MMTK-based interactive DomainFinder (Zoom in)
Characteristic features of biomolecular simulations that had to be taken into account are the long execution times of some simulation techniques (several weeks are not uncommon) and the complexity of the data structures describing biomolecules.
Choice of languages
The choice of Python plus C was made after an evaluation of various languages. I was rapidly convinced that only a mixture of a high-level interpreted language and a CPU-efficient compiled language could meet my seemingly conflicting requirements of rapid development and efficient execution.
For the high-level part, Tcl was ruled out because it could not handle the complex data structures required by the project. Perl was ruled out because of its unpleasant syntax (this was of course a subjective choice), and because of its badly integrated OO mechanism. Python scored high in readability, OO support, library support, and integration with compiled languages. Moreover, Numerical Python had just been released and was an important building block for my developments.
For the low-level part, Fortran 77 was eliminated because of its archaic character, lack of memory management, and portability issues in C-Fortran interfacing. C++ was a candidate, but ultimately not chosen because portability between compilers was still an issue in 1996, and because I considered the benefits of C++ for the small amount of compiled code in the project insufficient to compensate for the complexity of the language.
Library architecture
The architecture of MMTK is clearly Python-driven. To the user, it presents itself as a pure Python library. The C code in MMTK was written from scratch in the form of Python extension modules that only handle the few time-critical aspects: evaluation of interaction energies, and long-running iterative algorithms such as energy minimization and Molecular Dynamics, which run without any Python-related overhead. Extensive use is made of Numerical Python, LAPACK, and the netCDF library. MMTK provides multi-threading support for shared memory parallel machines, and MPI-based parallelization for distributed memory machines.
The biggest part of MMTK is a set of classes that describe atoms and molecules and manage a database of molecules and fragments. Biomolecules (proteins, DNA, and RNA) are handled by subclasses of the generic Molecule class. Another important subset of MMTK implements schemas for calculating interaction energies (called somewhat incorrectly "force fields" in the simulation community). I/O-related code is the third pillar of MMTK. It reads and writes a few popular file formats plus its own trajectory format that is based on the netCDF format. Contrary to other trajectory file formats, MMTK's netCDF files are both binary (and thus compact) files and portable between platforms. and moreover permit efficient access to nearly arbitrary subsets.
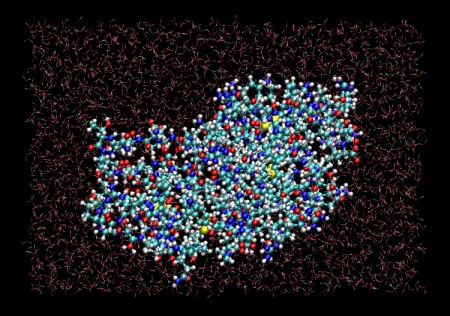
Snapshot from a Molecular Dynamics simulation of lysozyme in water, run with MMTK. Zoom in
Modularity and extendibility were important design criteria. Algorithms, energy terms, and specializations of the data types can be added without having to modify the MMTK code. The design of MMTK as a library, rather than a closed program, is essential for many applications.
An important aspect of biomolecular simulations is visualization. MMTK delegates this task to external tools. Two visualization programs, VMD and PyMOL, are particularly well integrated.
Most MMTK users access the library from simple Python scripts, but MMTK has also been used as a basis for end-user programs with graphical user interfaces, such as nMOLDYN and DomainFinder.
MMTK currently consists of about 18,000 lines of Python code, 12,000 lines of hand-written C code, and some machine-generated C code. The majority of the code was developed by one person during eight years as part of a research activity. Two modules, some functions, and many ideas were contributed by the user community.
Practical experience
MMTK and other Python libraries have been the basis for all my research projects for ten years. Many of these projects would not have been possible without the rapid prototyping that is characteristic for Python. In methodological work, development and testing time is essential: an idea that can be tried out in an afternoon will be tried out, whereas an idea that requires a week of work for evaluation is often put aside.
As with all open source projects, the size of the MMTK user community can only be estimated indirectly. The mailing list for MMTK users currently has 175 members, and the scientific publication that describes MMTK to computational chemists has been cited 30 times.
About the author
Konrad Hinsen is a researcher in theoretical physics working for the French Centre National de la Recherche Scientifique (CNRS). He was involved in the Numerical Python project and is the author of ScientificPython, a general-purpose library of scientific Python code.